
“中美双报”的背景
从注册申报的角度考虑,“中美双报”是指用同一套注册资料在中国和美国进行上市申报。在目前的实践中,一套资料同时适用于两地的注册申报并不现实。理想的情况是,申请人有一套通用的核心注册文件,仅需要做最低限度的调整,就能同时满足两地监管机构的要求。
而监管环境层面的改变可能是“中美双报”受到青睐的根本原因。随着2017年我国以成员国身份加入ICH后,全球法规监管框架越来越趋于一致,创新药物的跨地区开发有了坚实的法规基础。因此,“中美双报”的目的是在ICH法规框架下进行跨地域平行开发,并尽可能满足两个地区额外的法规要求。
对企业而言,开发创新药物需要大量的资金,企业迫切希望其开发的药物能够上市并获得利润,以支持后续的药物开发。在这样的背景下,美国作为全球创新药销售额最高的国家,自然是商业化的优先选择地域。同时,作为全球监管水平最高的国家,按照美国FDA的要求进行药物开发,不仅能支持当地的注册申请,也能为产品在其它区域进行申报奠定核心基础。
“中美双报”的可行性
如同之前所提及的,开发创新药物需要大量的资金,但是跨地域的药物开发所需资金成本更为高昂,且企业需要承担失败的风险。虽然“中美双报”可以加快药物的开发效率,其时间成本依然是一个不容忽视的问题。
在项目开发前期,对拟开发适应症在中美两地的实际临床需求进行调研是非常重要的。由于疾病存在一定的地域性特点,不同地区的发病率不尽相同,导致临床需求也完全不同。
另外,由于“中美双报”要求对两个区域的开发要求和法规有充分的理解,团队的中英沟通和撰写能力都不可或缺,这需要企业对自身团队能力进行评估,在必要时及时寻求经验丰富的外部团队提供支持。
“中美双报”的常见策略
合理利用政策加速药物研发
在竞争激烈的美国市场,对一些中小型企业而言,布局罕见病药物的开发,在满足患者的临床需求的同时,也能实现差异化竞争。罕见病指的是发病率极低的疾病,而治疗罕见病的药物被称为孤儿药。美国将每年患病人数小于20万人的疾病定义为罕见病。中国对于罕见病没有明确的定义,但也已发布了相应的罕见病目录供业界参考。
在美国,《孤儿药法案》以及《孤儿药现代化计划》在减免申请费用和部分税率,缩短审评时限,给予独占期等方面为开发者提供了政策上的倾斜,而我国同样在新版《药品注册管理办法》中规定罕见病药物可以申请优先审评,缩短审评时间。
利用快速审评通道提升开发效率
随着2020年最新版《药品注册管理办法》的颁布,我国监管机构已经提供了四条法定的药品加快上市注册程序,分别是突破性治疗药物程序、附条件批准程序、优先审评审批程序与特别审批程序。美国FDA同样也提供了Priority Review, Breakthrough Therapy, Accelerated Approval与Fast track项目,助力药物尽快上市,企业可以充分利用中美两国的这些快速审评途径来加快药品开发的效率。
值得注意的是,尽管两国提供的快速审评通道存在相似之处,但这并不意味着开发者的药物在某个国家被纳入快速审评通道后,另一方的监管机构也必定会同意将其纳入相似的通道。
开展国际多中心临床试验
美国是临床试验成本最高的国家,而中国除了成本优势,也因人口的数量较多,在招募病人等方面有一定的优势。为了提高开发效率并优化资源配置,国际多中心临床试验(MRCT)开始受到关注。
在过往的案例中,较为常见的是跨国企业的进口产品在关键验证性临床阶段开展国际多中心临床试验,待完成后将境外早期临床数据、桥接临床数据(如需)与国际多中心临床数据进行递交以寻求上市批准。而境内企业的产品出海时,也同样倾向于对境外的关键验证性临床数据进行补充,以满足递交要求。
而在“中美双报”同步开发的背景下,我们建议开发者在早期临床试验阶段就开展国际多中心临床试验,从而更早获得境内外临床数据,以提高临床开发效率。对于开发者而言,需要时刻注意两国之间临床实践的差异,比如两地临床开发的用时,监管机构对临床试验方案的考量,以及后续的沟通交流,甚至在临床申请资料撰写和递交过程的不同之处等方面尽早进行评估。
“中美双报”的注册要点
临床开发的启动用时
目前,我国临床试验申请批准时限为60个工作日,采用默示许可制度。而美国FDA的时限为30个自然日。获得临床试验批准后,后续的临床试验开展两国均需要进行伦理审查,研究合同签订,受试者招募等过程,此外,中国临床试验的开展还需要进行人遗审评。总体而言,我国在临床试验启动阶段的所需时间较长。

Pre-IND会议
中美监管机构均提供了临床申请前沟通交流会议(非强制性会议)的渠道,帮助申请人解决早期非临床/临床研究设计以及药学方面的问题,使得药品开发更有效率。尽管开发者能够通过该会议得到监管机构的重要科学性建议,但此过程往往需要数月的时间。
从监管机构的关注方面来看,FDA通常更加关注体外安全性数据,以确保首次人体试验的安全性。而CDE不仅关注安全性数据,也关注有效性数据,更会偏向预审查,以确保临床申请资料的质量。
监管机构的考量方向存在差异,但基于科学性数据的要求则是相同的。开发者与监管机构的沟通,需要基于企业对自身产品的深入研究,并提供扎实的支持性数据,以体现企业自身对问题的立场。

临床申请的撰写
现中美两国均为ICH的成员国,两个地区的药品注册相关技术要求已经获得了很大程度上的统一。因此,创新药物临床申请所需要提交的文件架构也趋于一致,按照ICH M4标准编写的通用技术文件(CTD)适用于两地的临床申请,其组织结构如下所示:
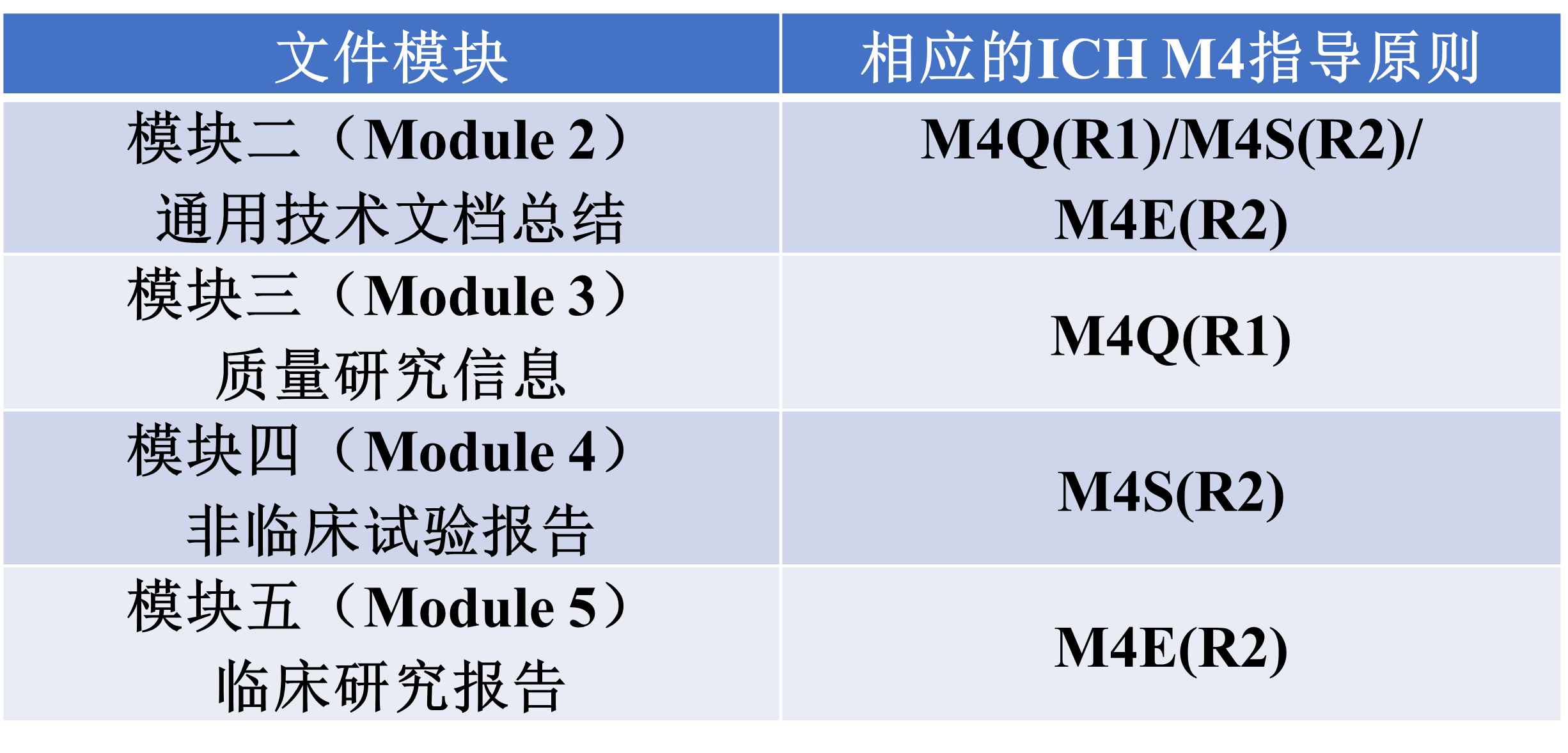
模块一为区域性要求,不属于CTD文件的一部分,通常需要遵循当地药品监管机构对于该章节编写的技术指导原则来准备。对于中国临床试验申请的模块一编写要求,国家药品监督管理局审评中心(CDE)在2020年7月1日颁布了《M4模块一行政文件和药品信息》。而美国临床试验申请模块一的结构可以参考FDA颁布的eCTD综合目录、标题和层级结构(Comprehensive Table of Contents Headings and Hierarchy)。此外,FDA也提供了IND准备与递交的指南文件(Investigational New Drug Applications Prepared and Submitted by Sponsor-Investigators),对模块一中需要提供的表格与文件做了解释说明。
上述信息主要适用于资料的组织架构,使得申报数据的呈现符合相应的格式要求。这并不包括开发者需要进行的研究种类,因为这部分需要考虑产品本身的特性,并参考ICH和当地监管机构所发布的指南。对于指南未覆盖的部分,需要基于科学逻辑判断可行性,并在必要时与监管机构进行沟通。
临床申请的递交
在递交临床申请前,两地一些差异化的要求需要提前准备,以避免延误。

*境内申请人递交FDA的申报文件时,需要关注该文件的可读性与专业性,以避免诸如用词不当,
语法错误,语序颠倒,表达方式不佳等翻译问题导致的IND审批暂停。
结语
“中美双报”是一个挑战,但也是一个机遇。随着全球药品监管环境的趋于统一,我国创新药物研发水平的不断提高,相信通过科学合理的策略和良好的注册项目管理与实践,国内企业出海的创新药将会越来越多。届时,“中美双报”也许将不会是一个特殊的“策略”,而是出于造福全球患者的考量。
发布于:2023年11月30日



