
美国食品和药物管理局 (FDA) 在药物开发过程中会通过一系列常规的正式会议为申办方提供许多互动和指导机会,包括在本系列中将会提到的A类、B类、B类(EOP)和 C 类会议(FDA指导原则:Formal Meetings Between the FDA and Sponsors or Applicants of PDUFA Products Guidance for Industry)。申办方应根据需要尽早与FDA进行互动,以确保双方就产品开发和申请策略方面达成一致,避免不必要的耗时或陷入代价高昂的麻烦中。
A类会议
A 类会议是针对推进已暂停的产品开发项目的恢复,或解决重要安全问题,可以包括以下类型:
- 21CFR10.75、312.48 和 314.103中提及的争端解决会议(FDA指导原则:Formal Dispute Resolution: Sponsor Appeals Above the Division Level)
- 讨论药物开发中的临床暂停情况,并着眼于讨论新的开发方向的会议
- 在收到 FDA 根据特殊临床方案评估程序的评估后,申办方要求召开的特殊临床方案评估会议。
- 在 FDA 做出监管行动(批准除外)的 3 个月内要求召开的 Post-action 会议
- 在 FDA 发出拒绝申请信函(refuse-to-file letter)后 30 天内要求召开的会议
B类会议
B 类会议是最常见的会议类型,与产品开发计划中的主要里程碑相关,包括以下类型:
- Pre-IND会议
- Pre-EUA会议
- Pre-NDA/ Pre-BLA会议
- 近期,FDA 还组织了一些用于审查综合疗效总结 (ISE) 和综合安全性总结 (ISS)的B 类会议。这些B类会议旨在协助申办方准备 ISS 和 ISE,通常被安排在预备会议之后和提交上市申请之前
- 在 FDA 做出监管行动(批准除外)3 个月以后要求召开的 Post-action 会议
- 在上市申请审查范围之外的,针对产品风险评估和应对策略或上市后要求的会议
- 讨论获得突破性疗法认证的产品的整体开发计划会议
B 类(EOP)会议是在药物开发已完成特定阶段、并准备进入下一个阶段时举行的会议,包括以下类型:
- End-of-phase 1(EOP1)会议,针对治疗危及生命和严重衰竭性疾病的生物制品(21 CFR 312 subpart E)和获得加速批准并用于严重/危及生命的的药物(21 CFR 314 Subpart H)
- End-of-phase 2 (EOP2) or Pre-phase 3 会议
C类会议
C 类会议则涵盖了 A 类、B 类或 B (EOP) 类会议未能囊括的有关产品开发和审查的主题的会议,包括以下类型:
- End-of-phase 2A (EOP2A) 会议
- OTC 各论反馈 (560FB) 会议
- Phase 4 (PF_4) 会议
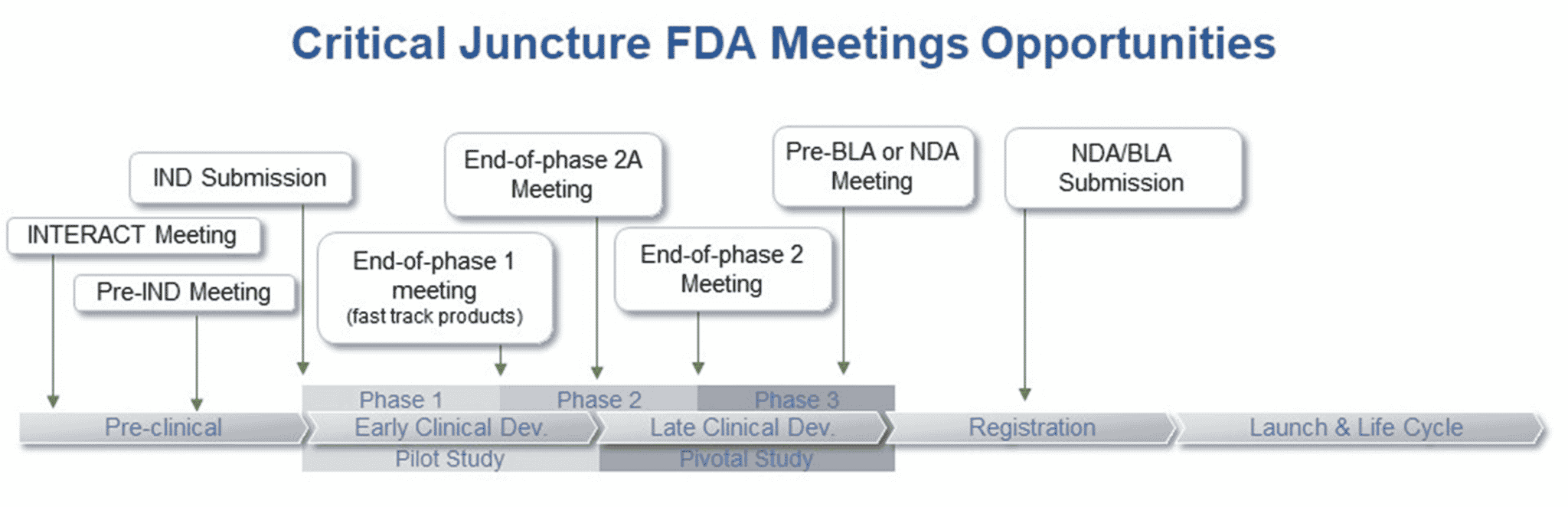
尽管在整个药物开发过程中,可能会进行数目繁多的会议,但是其中一些在开发关键节点召开的会议尤为重要:INTERACT、 pre-IND、 EOP1、 EOP2A、 EOP2和 Pre-NDA/Pre-BLA 会议。以下是这些会议的概述:
INTERACT 会议
INTERACT会议是创新类生物制品的申办方与 CBER 之间举行的非正式会议,旨在从 FDA 获得关于CMC、药理学/毒理学和/或临床开发计划方面的非约束性建议。
CBER 基于这些早期互动能够了解该创新性类产品的开发可能会带来与未知的安全性、复杂的制造工艺、技术和问题、创新性设备以及尖端检测方法的使用相关的独特挑战。
CDER目前不举行类似的会议,但在 2023 财年重新授权 PDUFA VII 时,这些会议将被启动。届时所有INTERACT 会议将成为正式的 PDUFA 会议。
Pre-IND 会议
Pre-IND会议是申办方与 CDER 或 CBER 之间举行的正式 PDUFA B 类会议。
在提交研究性新药申请 (IND) 之前,申办方可以要求召开此会议,以评估并就启动人体试验所需的动物研究设计以及 CMC 和临床方面与监管机构方面达成一致。Pre-IND会议并非在提交 IND 之前的必选项,但对于合理规划药物开发计划而言非常有价值,并且可以缩短总体上市时间。
在药物开发过程的早期阶段,FDA的主要职责之一是能为申办方提供指导。此外,无论是在Pre-IND会议期间,还是在审查 IND数据期间,FDA 的职责是确保被提交申请的产品足够安全用于启动相应的临床试验。
为了从与 FDA 的互动中获得最大收益,申办方必须明确他们的开发计划中存在的潜在问题,以期在计划的早期阶段中解决这些问题。此外,对临床方案的讨论,能够使 FDA 协助指导药物的开发过程。
EOP1 会议
EOP1会议是 IND 申办方与 CDER 或 CBER 之间举行的正式 PDUFA B 类会议。
只有用于治疗严重或危及生命的疾病的生物制剂(21 CFR 312 subpart E)和加速批准用于严重/危及生命的疾病的药物(21 CFR 312 subpart H)的申办方才有资格要求与 FDA 进行 EOP1 会议。
该会议的目标是审查第一阶段的临床研究,并就第二阶段的临床计划达成一致。这对于希望被纳入 NDA/BLA 加速审批通道的申办方来说尤为重要。
EOP2A 会议
EOP2A会议是 IND 申办方与 CDER 或 CBER 之间举行的正式 PDUFA C 类会议。
该会议为申办方与 FDA 进行深入的探索性讨论提供了机会,其重点是优化药物开发的接续步骤。
该会议的总体目的是讨论试验设计、建模策略和临床试验模拟方案的选项,以改进早期药物开发中暴露剂量-效应关系。
EOP2A 会议能够帮助下一阶段药物开发的给药方案选择,如何通过结合先前临床阶段中获得的定量知识并精心设计剂量-效应试验为后期临床试验提供支持。
EOP2 会议
EOP2或Pre-phase 3会议是 IND 申办方与 CDER 或 CBER 之间举行的正式 PDUFA B 类会议。
该会议能够帮助申办方最大限度地减少时间和成本方面的浪费,以及加快药物开发和评审过程。
EOP2 会议的目的是确定进入III期临床试验的安全性,评估III期临床计划和临床方案,以及当前研究的充分性、评估儿科安全性和进行有效的计划,并确定支持上市所需的任何其他信息能够被调用以满足审察要求。
尽管该会议并非必选项,但其对于药物开发计划的成功至关重要。有研究显示,对于选择进行EOP2 会议的申办方,其 NDA 获批率明显高于其他申办方。
Pre-NDA/Pre-BLA 会议
Pre-NDA/Pre-BLA会议分别是申办方与 CDER 或 CBER 之间举行的正式 PDUFA B 类会议。
该类会议对确保提交上市申请文件的良好组织和可读性而言至关重要。
Pre-NDA/Pre-BLA 会议的目的是确认从注册试验中获得的数据能够符合提交要求、发现任何尚未解决的重大问题、确认申办者所依赖的研究数据是充分且能用以确定药物的有效性、确认正在进行的需要进行的研究的状态能用以充分评估儿科安全性和有效性、向 FDA 审评人员介绍待提交的一般信息,以及讨论NDA/BLA中包含的数据最佳整理和呈现方式。
总结
在产品开发的整个过程中,申办方有许多与 FDA 之间互动的机会,其会议类型将取决于产品在开发过程中所处的阶段,以及请求 FDA 进行指导的内容。对于每种会议类型,其申请过程、申办方文件提供和时间线都有不同的要求。针对以上关键节点的重要会议类型,敬请期待本系列后续详细内容。
发布于:2022年7月21日




